L'amylose héréditaire ATTR: une maladie systémique potentiellement mortelle1-4
La maladie affecte plusieurs organes et entraîne des symptômes variables1,3,4
Étant donné que les fibrilles amyloïdes se déposent dans les tissus de l’ensemble de l’organisme, y compris les nerfs, le cœur ou le système gastro-intestinal (GI), les patients atteints d’amylose hATTR peuvent présenter un spectre de symptômes sensitifs, moteurs, autonomes et cardiaques.1-5
En réalité, plus de la moitié des patients atteints d’amylose hATTR présentent un phénotype mixte.6,7
Ensemble des signes et symptômes possibles de l’amylose hATTR
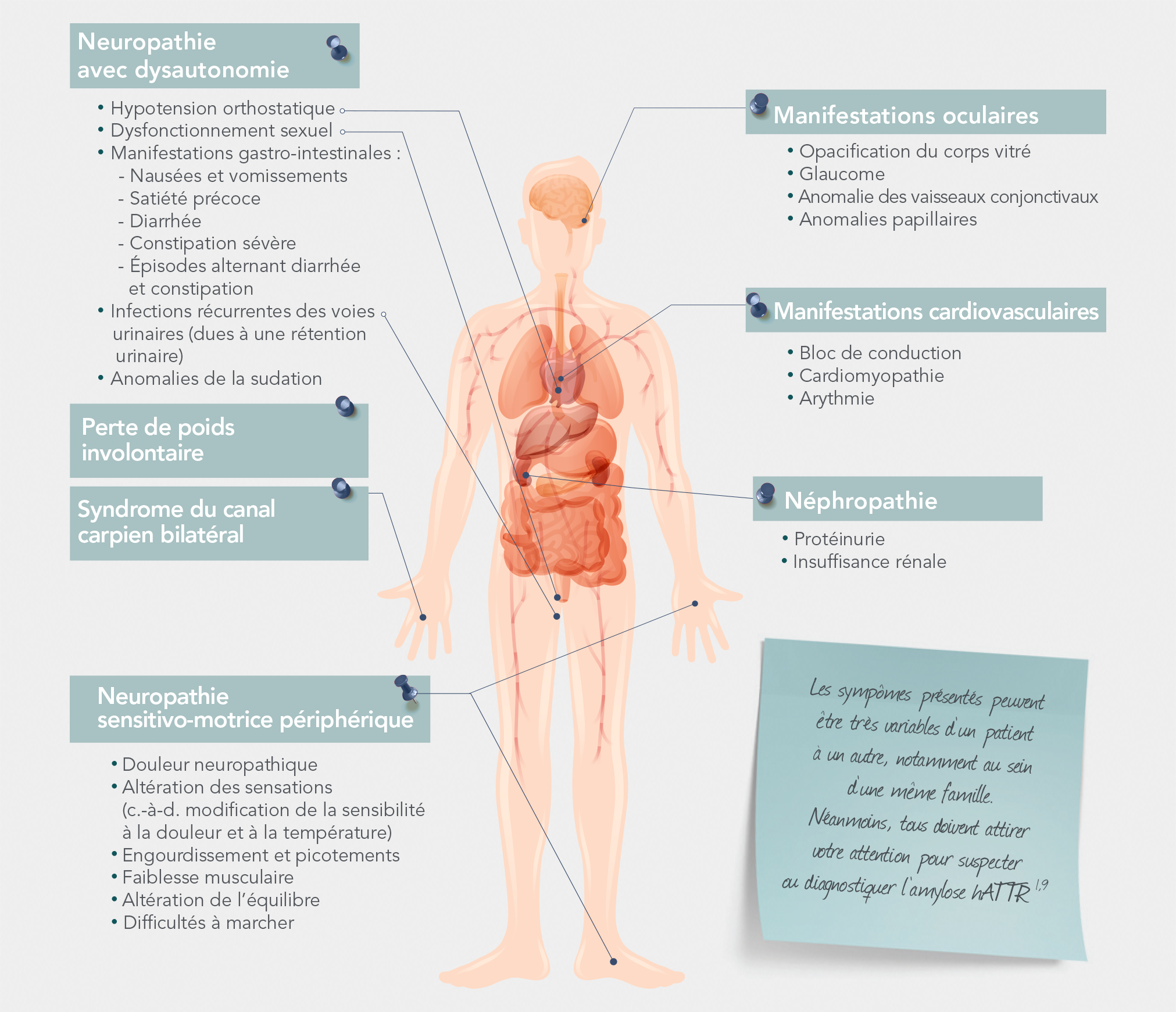
Adapté d’après Conceição I, et al. J Peripher Nerv Syst. 2016;21(1):5-9.
La présentation des symptômes varie considérablement selon les patients, avec mutation. Les symptômes peuvent également varier entre des patients de la même famille8
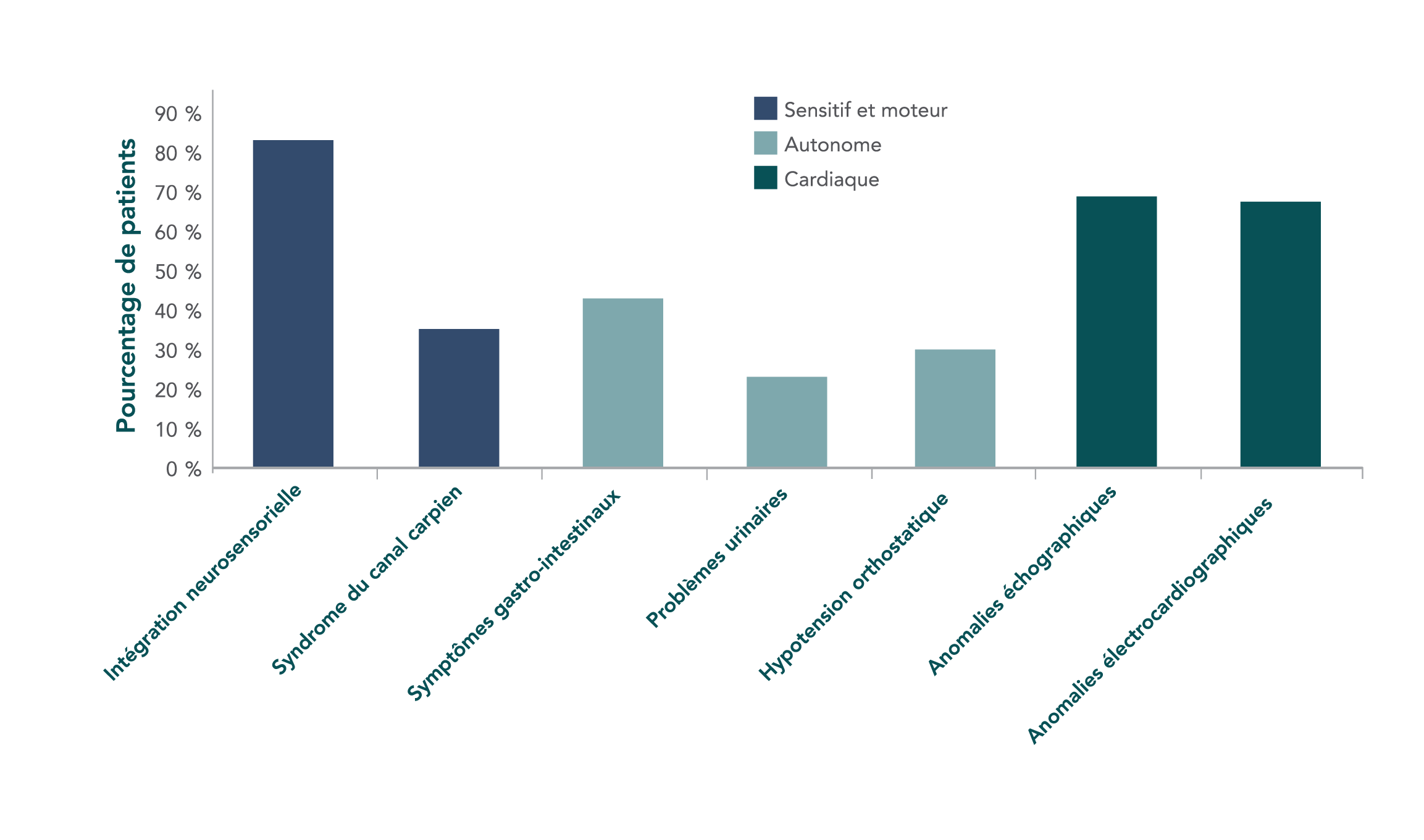
Caractéristiques cliniques de base de 186 personnes atteintes d’amylose ATTR héréditaire, au cours d’une étude multicentrique.6