L'amylose héréditaire ATTR : une maladie familiale évolutive et potentiellement mortelle1-3
Qu’est-ce que l’amylose hATTR ?
L’amylose hATTR est une maladie héréditaire autosomique dominante, systémique, évolutive et qui engage le pronostic vital. Elle est provoquée par une mutation du gène de la TTR. Les fibrilles amyloïdes sont déposées dans plusieurs sites de l’organisme, notamment les nerfs, le cœur et le tractus gastro-intestinal, où elles provoquent des lésions à l’origine des symptômes cliniques2,4
Formation des fibrilles amyloïdes2,4,5
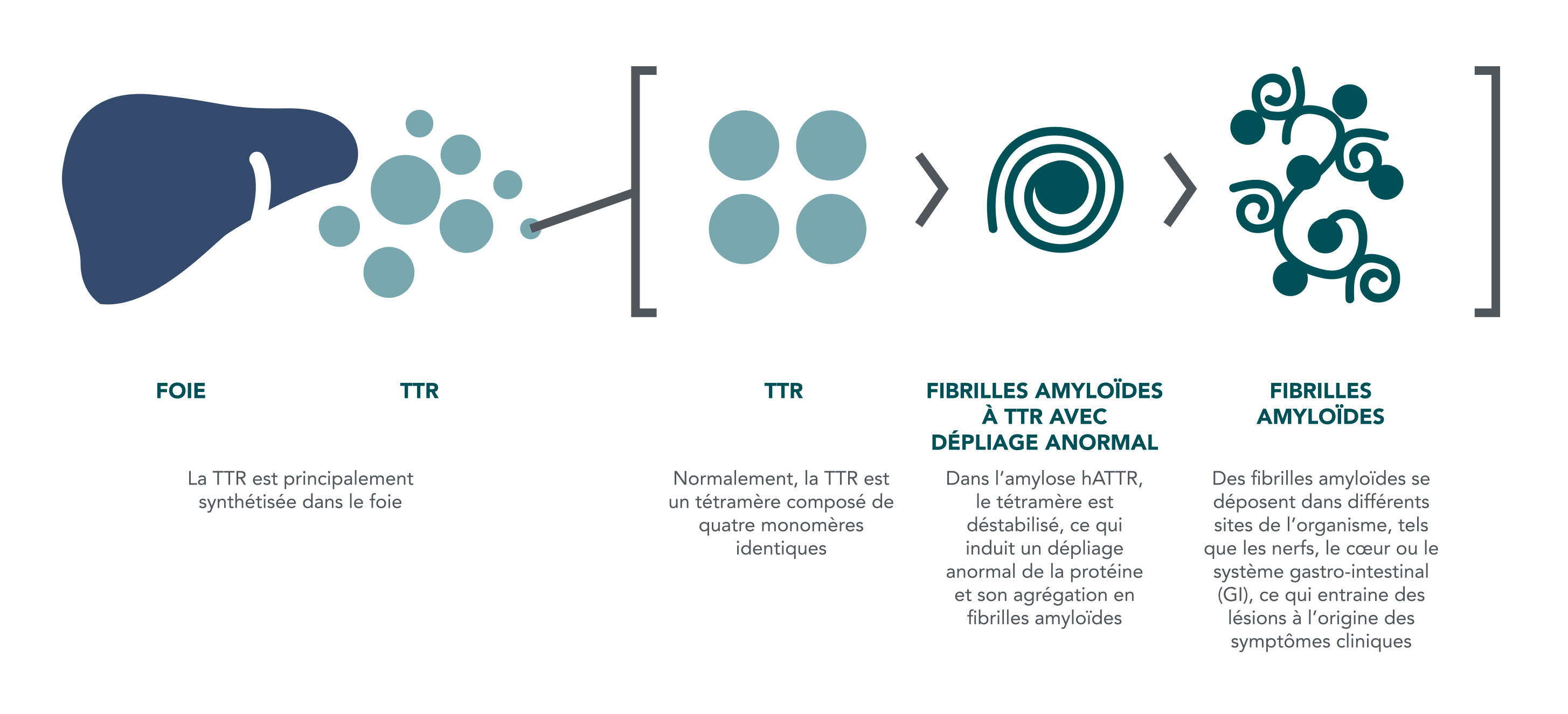
Références :
- Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675-682.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50-57.
- Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528-540.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625-638.
- Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036-1043.
- Ando Y, Coelho T, Berk JL, et al. Guidelines of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
- Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520-528.
- Adams D, Gonzalez-Duarte A, O’Riordan W, et al., an investigational RNAi therapeutic for the treatment of hereditary ATTR amyloidosis with polyneuropathy: baseline demographics from the phase 3 APOLLO study. In: The XVth International Symposium on Amyloidosis. Uppsala, Sweden: ISA International Society of Amyloidosis; July 3-7, 2016. PA 82.
- Coelho T, Maurer MS, Suhr OB. THAOS—The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63-76.
- Castaño A, Drachman BM, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178.